

RNA Dysregulation in Neurodegeneration
Overview of Bhinge Lab research at the Living Systems Institute, University of Exeter

RNA dysregulation has emerged as a central theme in the pathogenesis of many neurodegenerative disorders with mutations in several RNA-binding proteins deemed to be directly involved in causing neuronal death. We are interested in how RNA regulation contributes to neuronal homeostasis and how its dysregulation leads to disease. The life of an RNA molecule is regulated at multiple levels. For example, transcription factors and the epigenetic machinery control transcriptional output, RNA-binding proteins control RNA splicing and trafficking, while microRNAs fine-tune translational output. Each of these players interact in a complex network that is cell-type specific to effect neuronal homeostasis. We believe that disruption of these cell-type specific networks leads to the differential susceptibility observed in neurodegenerative conditions. For example, Amyotrophic lateral sclerosis (ALS) is characterised by a preferential loss of motor neurons whereas certain cortical neuronal subtypes are affected in Fronto-temporal dementia (FTD) and Alzheimer's disease (AD).
We use neurons differentiated from patient-derived and genome-edited pluripotent stem cells to develop in vitro human models of ALS, FTD and AD. These models are further investigated using a combination of single-cell RNA-Seq, epigenomics and functional genomics screens to identify therapeutic targets that can halt the relentless neuronal loss in these diseases.
CURRENT PROJECTS
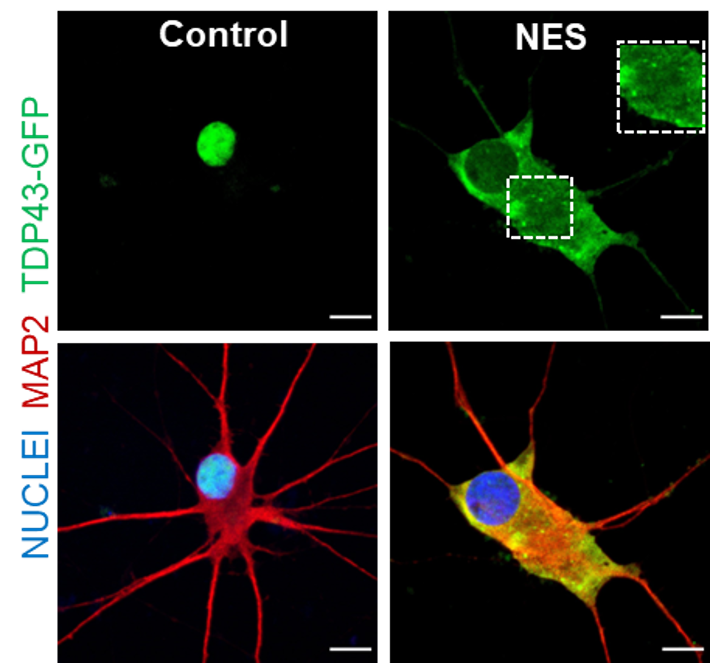
RNA dysregulation due to TDP43 mislocalisation in motor neurons, cortical neurons and oligodendrocytes.
Neuromuscular junction defects due to TDP43 mislocalisation.
Disruption of microRNA networks in FUS and sporadic ALS.
Unbiased mapping of RNA-binding protein targets across the transcriptome due to TDP43 proteinopathy.
Epigenetic mechanisms in ALS.
Optical genetic screens to identify genetic drivers of TDP43 mislocalisation.
Repurposing clinically approved drugs for ALS.
A human model of TDP43 proteinopathy
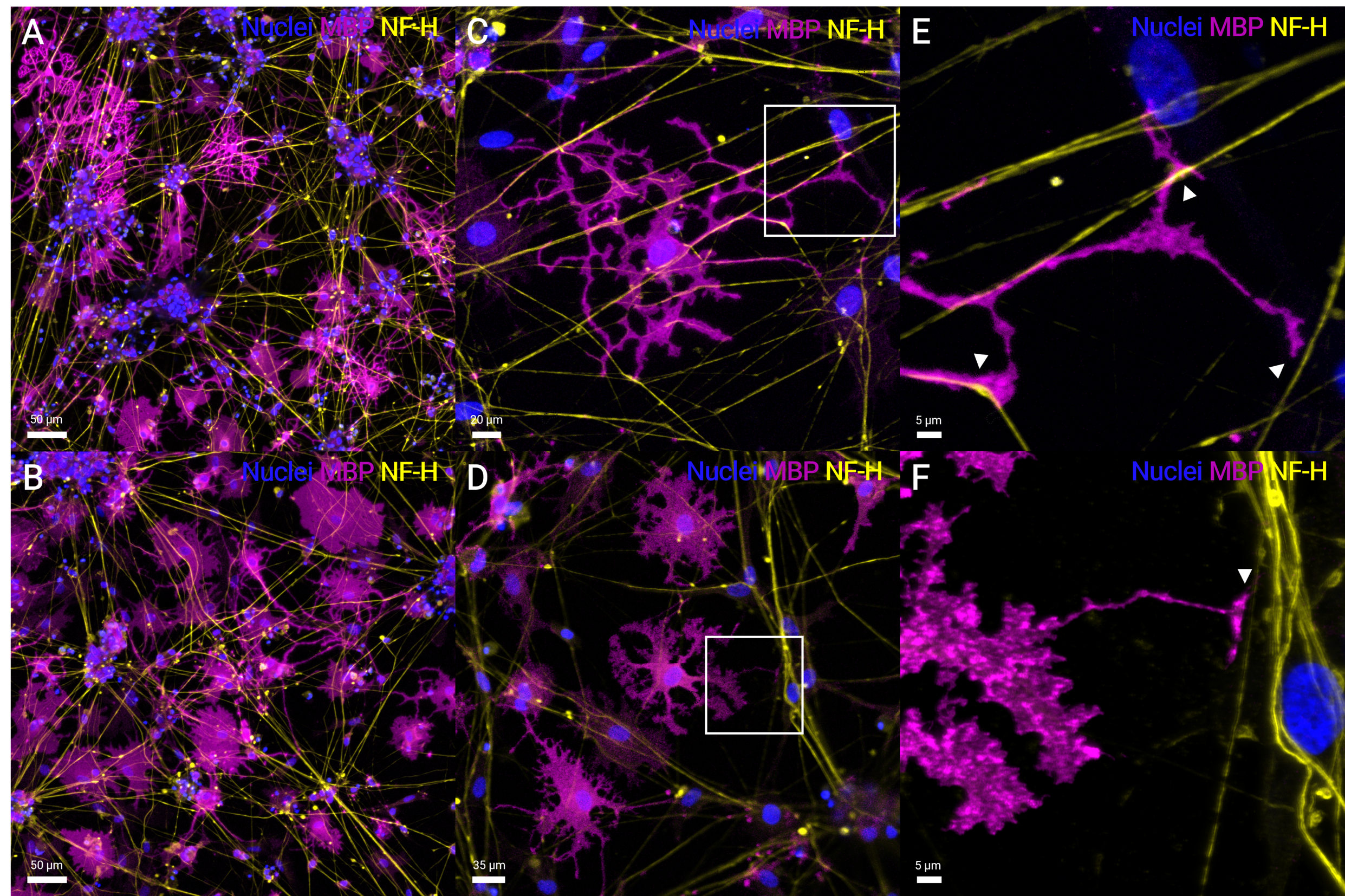
Human iPSC-derived oligodendrocytes myelinating motor neuron axons
Human iPSC-derived skeletal muscle stained with MYHC
Human MN-muscle assembloids
Motor neurons stained with Neurofilament-M (green) and ISL1 (magenta)
Sensory neurons self-organising into clusters in long-term neuronal cultures


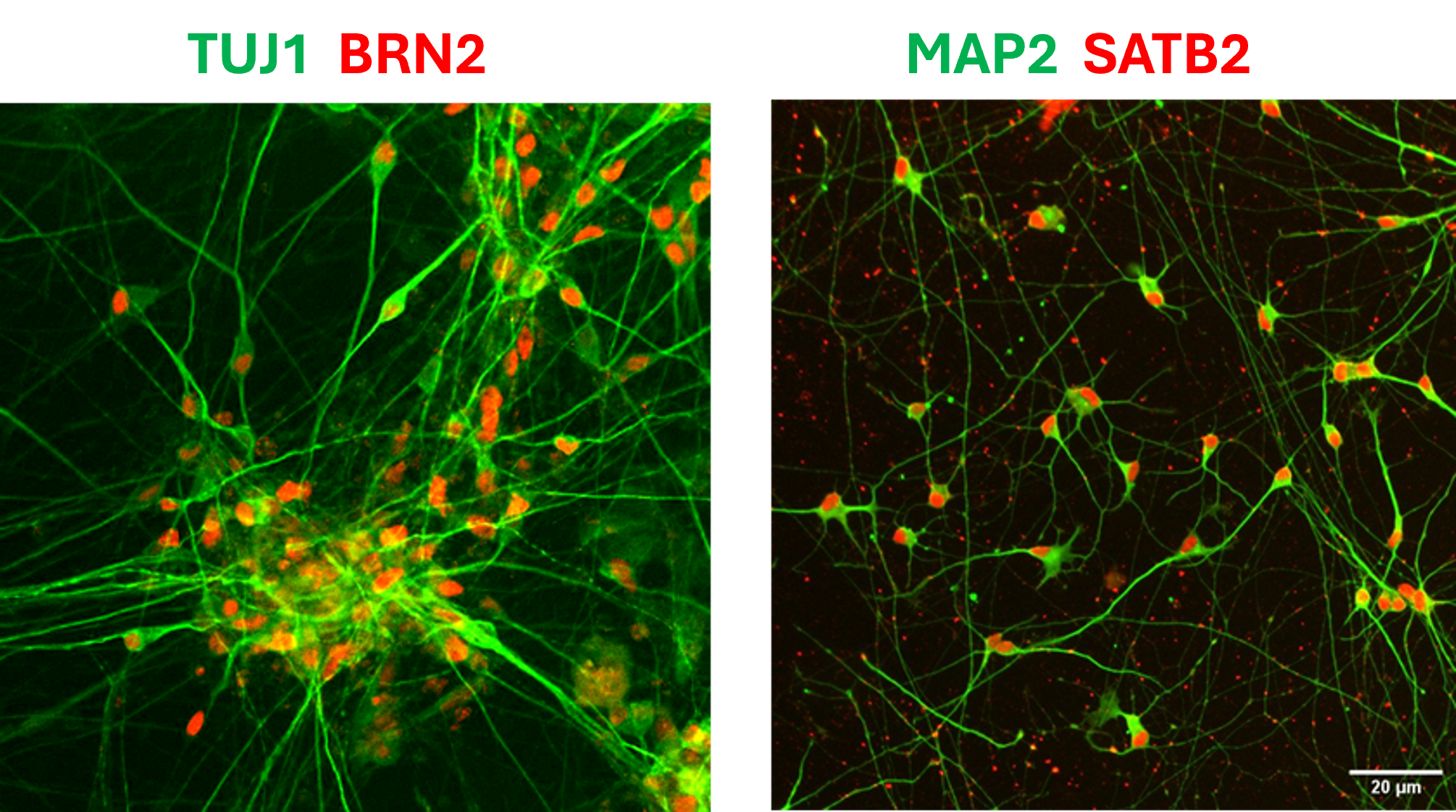
Human iPSC-derived cortical neurons
Hippocampal neurons stained with PROX1 (green) and MAP2 (magenta)

Motor neurons firing in response to potassium
EXTERNAL COLLABORATORS
Prof. Siddharthan Chandran, The University of Edinburgh, UK
Prof. Jernej Ule, The Francis Crick Institute, London, UK
Dr. Bhuvaneish T Selvaraj, The University of Edinburgh, UK
Dr. Johnathan Cooper-Knock, University of Sheffield, UK
Prof. Gene Yeo, University of California San Diego, USA
Prof. Kevin Talbot, University of Oxford, UK
Prof. Andrew Yoo, Washington University in St. Louis, USA
Dr. Asif Javed, The University of Hong Kong
Assoc. Prof. Olaf Ansorge, University of Oxford, UK
Prof. Aaron Gitler, Stanford University, USA
Assoc. Prof. Alessandro Rosa, Sapienza University of Rome, Italy
Dr. Jeremie Poschmann, INSERM, Université de Nantes, France
FUNDING
